Docker怎么实现Samtools截取基因组序列
发布于 2022-03-19 21:10:24
上一篇:MD5的概念是什么 下一篇:ajaxSuccess使用注意事项是什么
目录
推荐阅读
-
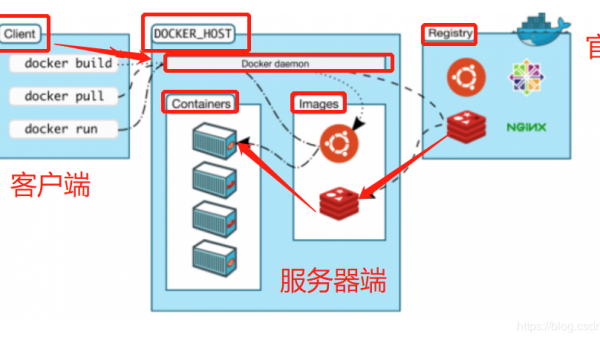
Linux怎么安装和使用Harbor搭建容器镜像仓库
-
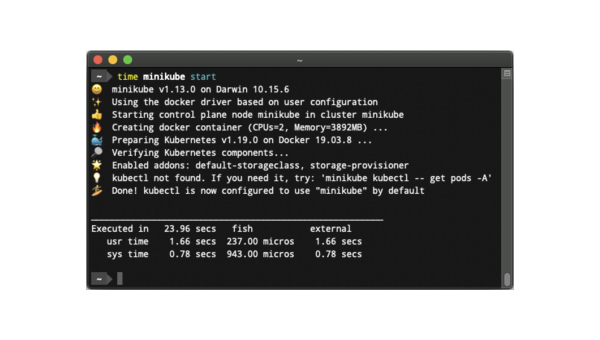
Linux怎么安装和使用Minikube搭建Kubernetes集群
-
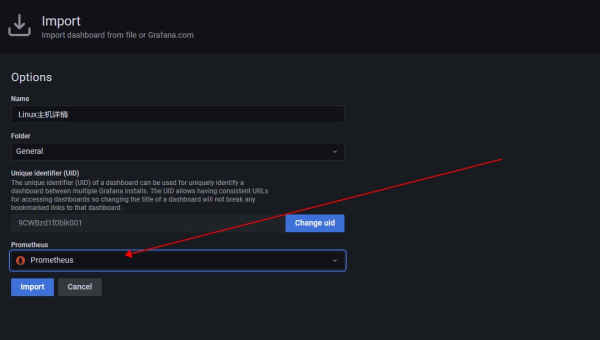
Linux如何安装和配置GrafanaLoki日志聚合系统
-
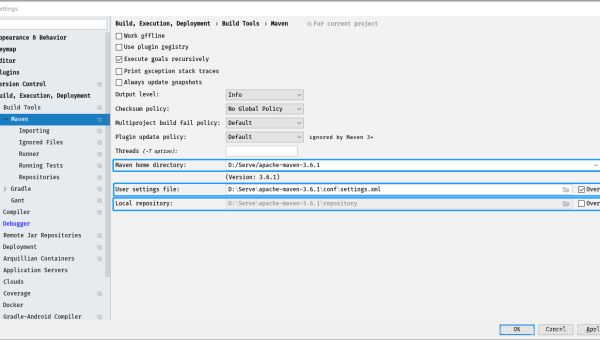
IntelliJ IDEA Docker 集成:微服务本地调试与镜像构建
-
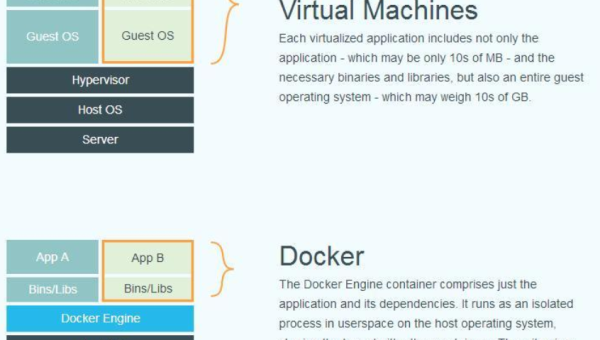
Docker 数据卷管理:持久化存储与容器间数据共享
-
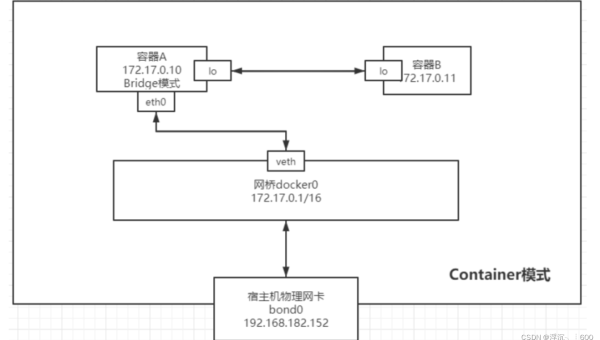
Docker 网络模式对比:bridge、host、none 模式的适用场景
-
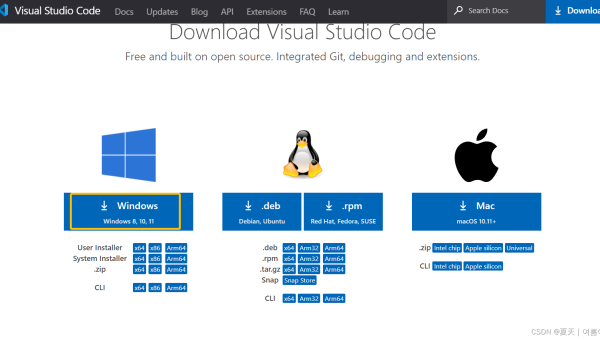
VS Code Remote Containers:基于 Docker 的跨环境开发配置全流程
-
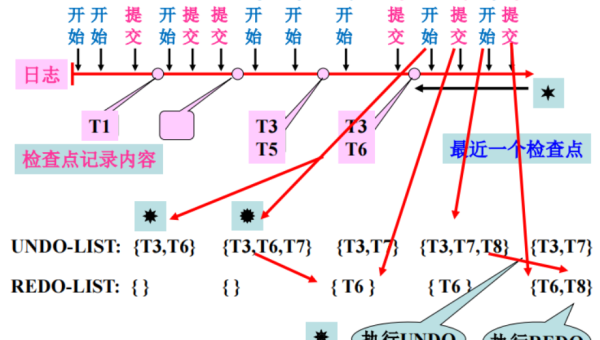
Docker 镜像分层原理:UnionFS 机制与缓存利用深度解析
-
Docker 内容信任机制:分布式系统中镜像完整性验证
-
Docker 多阶段构建优化:减少镜像体积与依赖包清理